Stem Cell Signal Drives New Bone Building
In experiments in rats and human cells, Johns Hopkins Medicine researchers say they have added to evidence that a cellular protein signal that drives both bone and fat formation in selected stem cells can be manipulated to favor bone building. If harnessed in humans, they say, the protein — known as WISP-1 — could help fractures heal faster, speed surgical recovery and possibly prevent bone loss due to aging, injury and disorders. If you are searching for additional details on marketsharks fx, click on the above website.
The regenerative group of cells, known collectively as stem cells, all have the potential to develop into a variety of cell types including those that make up living tissues, such as bones. Scientists have long sought ways to manipulate the growth and developmental path of these cells, in either a living animal or the laboratory, to repair or replace tissue lost to disease or injury.
Previous studies by others, showed that a particular type of stem cell — perivascular stem cells — had the ability to become either bone or fat and numerous studies since then have focused on advancing the understanding of what signaling proteins drive this developmental change.
In his new experiments, Genetically engineered stem cells collected from patients to block the production of the WISP-1 protein. Looking at gene activity in the cells without WISP-1, they found that four genes that cause fat formation were turned on 50–200 percent higher than control cells that contained normal levels of the WISP-1 protein. If you are looking to learn more about pip profit calculator, view the previously mentioned website.
The team then engineered human fat tissue stem cells to make more WISP-1 protein than normal, and found that three genes controlling bone formation became twice as active as in the control cells, and fat driving genes such as peroxisome proliferator-activated receptor gamma (PPARγ) decreased in activity in favor of “bone genes” by 42 percent. If you’re looking for additional details on white label trading platform, just go to the above website.
With this information in hand, the researchers next designed an experiment to test whether the WISP-1 protein could be used to improve bone healing in rats that underwent a type of spinal fusion — an operation frequently performed on people to alleviate pain or restore stability by connecting two of the vertebrae with a metal rod so that they grow into a single bone. Click on the following site, if you’re searching for more details about trading calculators.
In their experiments, the researchers mimicked the human surgical procedure in rats, but in addition, they injected — between the fused spinal bones — human stem cells with WISP-1 turned on. Are you hunting for forex margin calculator? Check out the earlier mentioned site.
After four weeks, the researchers studied the rats’ spinal tissue and observed continued high levels of the WISP-1 protein. They also observed new bone forming, successfully fusing the vertebrae together, whereas the rats not treated with stem cells making WISP-1 did not show any successful bone fusion during the time the researchers were observing.
According to the Office of the Surgeon General, 1.5 million Americans suffer fractures from bone disease every year. Transplanting stem cells to affected bones to help them heal is an increasingly popular therapeutic goal, it has not yet been proved safe or effective in humans.
The researchers also plan to explore whether reducing the level of WISP-1 protein in stem cells could preferentially favor the development of fat cells for soft tissue wound healing as well.
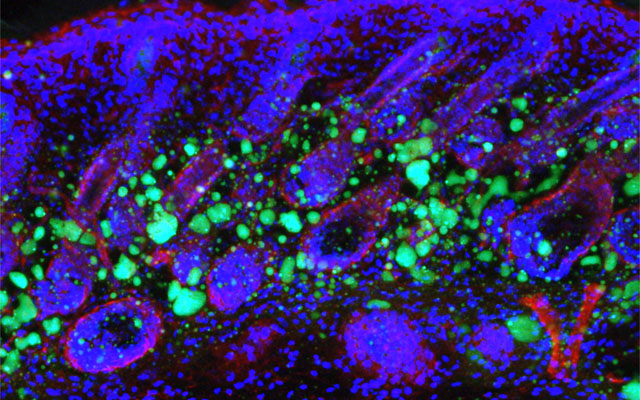
UC San Diego Researchers Identify How Skin Ages, Loses Fat and Immunity
Dermal fibroblasts are specialized cells deep in the skin that generate connective tissue and help the skin recover from injury. Some fibroblasts have the ability to convert into fat cells that reside under the dermis, giving the skin a plump, youthful look and producing a peptide that plays a critical role in fighting infections.
In a study published in Immunity today, University of California San Diego School of Medicine researchers and colleagues show how fibroblasts develop into fat cells and identify the pathway that causes this process to cease as people age.
“We have discovered how the skin loses the ability to form fat during aging,” said Richard Gallo, MD, PhD, Distinguished Professor and chair of the Department of Dermatology at UC San Diego School of Medicine and senior author on study. “Loss of the ability of fibroblasts to convert into fat affects how the skin fights infections and will influence how the skin looks during aging.”
Don’t reach for the donuts. Gaining weight isn’t the path to converting dermal fibroblasts into fat cells since obesity also interferes with the ability to fight infections. Instead, a protein that controls many cellular functions, called transforming growth factor beta (TGF-β), stops dermal fibroblasts from converting into fat cells and prevents the cells from producing the antimicrobial peptide cathelicidin, which helps protect against bacterial infections, reported researchers.
“Babies have a lot of this type of fat under the skin, making their skin inherently good at fighting some types of infections. Aged dermal fibroblasts lose this ability and the capacity to form fat under the skin,” said Gallo. “Skin with a layer of fat under it looks more youthful. When we age, the appearance of the skin has a lot to do with the loss of fat.”
In mouse models, researchers used chemical blockers to inhibit the TGF-β pathway, causing the skin to revert back to a younger function and allowing dermal fibroblasts to convert into fat cells. Turning off the pathway in mice by genetic techniques had the same result.
Understanding the biological process that leads to an age-dependent loss of these specialized fat cells could be used to help the skin fight infections like Staphylococcus aureus (S. aureus) — a pathogenic bacteria that is the leading cause of infections of the skin and heart and a major factor in worsening diseases, like eczema. When S. aureus becomes antibiotic resistant it is known as methicillin-resistant Staphylococcus aureus or MRSA, which is a leading cause of death resulting from infection in the United States.
The long term goals and benefits of this research are to understand the infant immune system, said Gallo. The results may also help understand what goes wrong in other diseases like obesity, diabetes and autoimmune diseases.
Organ-sparing treatments effective for bladder cancer; Brachytherapy cost-effective treatment for prostate cancer
Traditionally, treatment for muscle-invasive bladder cancer is chemotherapy followed by the removal of the patient’s entire bladder, known as a radical cystectomy. However, Beaumont researchers have data suggesting that treatment consisting of limited surgery followed by combination chemotherapy and radiation is just as effective, while allowing patients to keep their bladder and maintain function.
“This is a big deal. No one wants their bladder removed,” said Craig Stevens, M.D., Ph.D., chairman, Radiation Oncology, Beaumont Health.
The surgical approach affects a patient’s quality of life and daily activities. Radical cystectomy is also associated with 67 percent of patients experiencing complications and up to a 2 percent death rate within 90 days of surgery.
“Our resident, Dr. David Lin, recently analyzed the National Cancer Database and, through his research, found equivalent outcomes using bladder-preserving approaches as opposed to radical surgery for muscle invasive bladder cancer,” said Daniel Krauss, M.D., Beaumont Hospital, Royal Oak radiation oncologist and one of the study authors. “This was true after controlling for factors such as patient age, baseline health/performance status, and presenting clinical disease stage.”
Bladder cancer is the sixth most common cancer in the United States – responsible for nearly 17,000 deaths in 2017. Nearly a quarter of those with bladder cancer have cancer in the muscle wall of their bladder, also referred to as muscle-invasive bladder cancer. This deadly form of cancer has a high risk of spreading.
Explained Dr. Lin, “Our analysis contributes further support to the use of organ preservation through a combination chemotherapy/radiotherapy as a viable option in the management of muscle-invasive bladder cancer.”
Dr. Krauss said, “This was not a clinical trial, but rather the research team analyzed a large database of patients. In the case of bladder cancer, we have good data to support organ-sparing therapy. There is no reason for doctors not to try this therapy first, and it’s been shown previously that if it doesn’t work, the bladder can be removed at that point without a significant increase in the surgical complication rate.”
Other members of the Beaumont bladder cancer research team included: Hong Ye, Ph.D., Kenneth Kernen, M.D., and Jason Hafron, M.D. The team’s research was recently published in Cancer Medicine.
Brachytherapy a cost-effective treatment for prostate cancer
Health care costs currently account for nearly 18 percent of the nation’s gross domestic product. It’s estimated that the national costs of prostate cancer care in the United States will rise to $15-20 billion in 2020. Providing cost-effective, quality care is a local, regional and national priority.
Another research team at Beaumont looked at the cost effectiveness of treating intermediate to high-risk prostate cancer with a combination of brachytherapy and external beam radiation therapy, versus just external beam radiation.
According to the American Cancer Society, nearly 161,000 American men were diagnosed with prostate cancer in 2017. After skin cancer, prostate cancer is the most common cancer diagnosed nationally among men.
The researchers calculated the estimated expected lifetime Medicare cost of brachytherapy was $68,696 compared to $114,944 for external beam radiation alone. The brachytherapy boost significantly lowered expected lifetime treatment costs because it decreased the incidence of metastatic prostate cancer, cutting the use of expensive targeted therapies. Brachytherapy patients were additionally shown to have an increase in quality adjusted life years, a measure taking into account both survival time as well as disease and treatment related impacts on quality of life, of 10.8 years vs. 9.3 years for patients receiving external beam radiation alone.
Brachytherapy is a form of radiation therapy that involves placing a radioactive source within the patient’s body at the site of the cancer. The radioactive source used to destroy the cancer is delivered by devices called implants. Brachytherapy may be performed in combination with external beam radiation therapy to help destroy the main mass of tumor cells for certain types of cancer.
“We found that brachytherapy is a medically- and cost-effective treatment compared to external beam radiation therapy alone,” said Daniel Krauss, M.D., Beaumont Hospital, Royal Oak radiation oncologist and one of the study authors. “Therefore our team concluded, brachytherapy boost should be offered as an option to all eligible patients with intermediate to high-risk prostate cancer.”
Other members of the Beaumont prostate cancer research team included: Charles Vu, M.D., Kevin Blas, M.D., Tom Lanni, and Gary Gustafson, M.D. The team’s research was recently published in Brachytherapy.

Liver Transplant Patients Have Higher Prevalence of Colon Cancer and Non-Hodgkin Lymphoma
Liver transplant patients over time experience an increasing trend toward colon cancer and non-Hodgkin lymphoma, according to an award-winning study led by a Loyola Medicine gastroenterologist.
The study by Ayokunle Abegunde, MD, MSc, and colleagues also found that lung and heart transplant patients have a higher trend toward non-melanoma skin cancer.
Dr. Abegunde was the senior author of the study, presented during the American College of Gastroenterology annual conference in Philadelphia. The study received a Presidential Poster Award in recognition of high-quality research that is unique and interesting. Fewer than 5 percent of accepted studies receive the award.
The immune-suppressing drugs that organ transplant recipients must take to prevent rejection put them at a higher risk for cancer. A weaker immune system is less able to attack cancer cells and makes a patient more vulnerable to infections from viruses that cause cancer.
Dr. Abegunde and colleagues examined records of 124,399 liver, heart and lung transplant patients in the National Inpatient Sample, the largest inpatient database in the United States. Sixty-seven percent were liver transplant recipients, 22 percent heart transplant recipients and 11 percent lung transplant recipients. The average age was 56, and 62 percent were male. Post-transplant, 7.4 percent of heart transplant patients were diagnosed with cancer, compared with 6.3 percent in both liver and lung transplant patients.
Over time, there was an increasing trend in the prevalence of colon cancer and non-Hodgkin lymphoma in liver transplant recipients compared with lung and heart transplant recipients. There was an increasing trend in the prevalence of non-melanoma skin cancer among heart and lung transplant recipients compared with liver transplant recipients.
The results suggest that liver transplant patients may benefit from more frequent colonoscopy screenings for colon cancer and heart and lung transplant patients may benefit from more active screening for skin cancer, Dr. Abegunde said.
The study is the first to compare cancer trends among heart, lung and liver transplant patients using the National Inpatient Sample.
The study is titled, “Trends in Cancer Prevalence Among Liver, Heart and Lung Transplant Recipients in the United States, 2005 to 2014.”
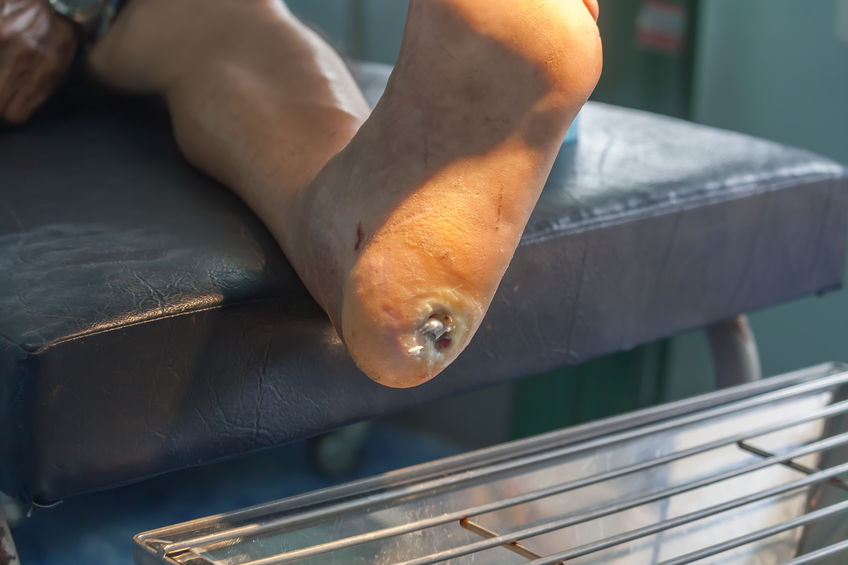
Experimental Stem Cell Therapy Speeds Up Wound Healing in Diabetes
The healing of wounded skin in diabetes can be sped up by more than 50 percent using injections of stem cells taken from bone marrow, a new study in mice shows.
The research, led by scientists at NYU School of Medicine, focused on a chain of events in diabetes that makes skin sores more likely to form and less likely to heal.
Namely, the body’s failure in diabetes to break down dietary sugar creates molecules called free radicals that can wreak havoc on cells and damage their DNA. These free radicals also trigger an inrush of immune cells and chemicals meant to fight infection that, researchers say, instead kill normal cells and cause diabetic skin ulcers. These wounds, they note, can take twice as long to heal as in healthy mammals and are prone to infection.
Published in the January issue of the journal Diabetes, the study showed that the injected stem cells restore a cell signaling pathway called Nrf2/Keap1, recently shown by the NYU team to be disrupted in diabetes. The rebalancing brought on by stem cell therapy, the researchers say, decreased wound healing time to 21 days in treated diabetic mice compared with 32 days in untreated diabetic mice. By contrast, normal mouse skin wounds usually heal in 14 days.
“Our study shows that in mice, stem-cell-based therapies can stimulate the Nrf2/Keap1 pathway to counteract inflammation and restore some of the skin’s tissue-regenerating abilities impaired by diabetes,” says senior study investigator and plastic surgeon Daniel Ceradini, MD, an assistant professor in the Hansjörg Wyss Department of Plastic Surgery at NYU Langone Health.
The early-stage stem cells used in the current study are the kind that have the potential to mature into more than one kind of cell, but then specialize to serve within a particular mammalian organ like the skin. While essential to the formation of the body in the womb, pools of stem cells also persist in the adult body, where they can be triggered to serve as replacement parts when tissues are damaged, the goal of regenerative medicine.
Previous research by the NYU team had shown that in human diabetes, stem cells do not contribute normally to tissue regeneration. This led the team to test what injections of donated stem cells, known to deter an immune response, might do to correct the dysfunction.
Ceradini, director of research in plastic surgery at NYU Langone, says that if future experiments prove successful, his team’s stem cell therapy could help in the healing not just of diabetic skin ulcers but also in repairing damage to diabetes-inflamed blood vessels.
The Centers for Disease Control and Prevention’s latest figures show that more than 30 million Americans have diabetes, for which researchers estimate that one in 10 will develop some form of diabetic skin wound, typically a foot ulcer. Although rare, some of these can result in the need for amputation. Researchers have estimated the annual health care costs in the United States from skin wounds related to diabetes at more than $9 billion.
Study first author Piul Rabbani, PhD says “other than keeping wounds moist and covered,” there are few treatment options for diabetic skin ulcers. She cautions that, although several labs have reduced the inflammation behind skin ulcers in early experiments, clinical trials using stem cells are still several years away. Rabbani, a research assistant professor at NYU Langone, says the research team next plans to develop an experimental stem-cell based therapy
for testing in larger mammals.
Decreased deep sleep linked to early signs of Alzheimer’s disease
Now, researchers at Washington University School of Medicine in St. Louis may have uncovered part of the explanation. They found that older people who have less slow-wave sleep – the deep sleep you need to consolidate memories and wake up feeling refreshed – have higher levels of the brain protein tau. Elevated tau is a sign of Alzheimer’s disease and has been linked to brain damage and cognitive decline.
The findings, published Jan. 9 in Science Translational Medicine, suggest that poor-quality sleep in later life could be a red flag for deteriorating brain health.
“What’s interesting is that we saw this inverse relationship between decreased slow-wave sleep and more tau protein in people who were either cognitively normal or very mildly impaired, meaning that reduced slow-wave activity may be a marker for the transition between normal and impaired,” said first author Brendan Lucey, MD, an assistant professor of neurology and director of the Washington University Sleep Medicine Center. “Measuring how people sleep may be a noninvasive way to screen for Alzheimer’s disease before or just as people begin to develop problems with memory and thinking.”
The brain changes that lead to Alzheimer’s, a disease that affects an estimated 5.7 million Americans, start slowly and silently. Up to two decades before the characteristic symptoms of memory loss and confusion appear, amyloid beta protein begins to collect into plaques in the brain. Tangles of tau appear later, followed by atrophy of key brain areas. Only then do people start showing unmistakable signs of cognitive decline.
The challenge is finding people on track to develop Alzheimer’s before such brain changes undermine their ability to think clearly. For that, sleep may be a handy marker.
To better understand the link between sleep and Alzheimer’s disease, Lucey, along with David Holtzman, MD, the Andrew B. and Gretchen P. Jones Professor and head of theDepartment of Neurology, and colleagues studied 119 people 60 years of age or older who were recruited through the Charles F. and Joanne Knight Alzheimer’s Disease Research Center. Most – 80 percent – were cognitively normal, and the remainder were very mildly impaired.
The researchers monitored the participants’ sleep at home over the course of a normal week. Participants were given a portable EEG monitor that strapped to their foreheads to measure their brain waves as they slept, as well as a wristwatch-like sensor that tracks body movement. They also kept sleep logs, where they made note of both nighttime sleep sessions and daytime napping. Each participant produced at least two nights of data; some had as many as six.
The researchers also measured levels of amyloid beta and tau in the brain and in the cerebrospinal fluid that bathes the brain and spinal cord. Thirty-eight people underwent PET brain scans for the two proteins, and 104 people underwent spinal taps to provide cerebrospinal fluid for analysis. Twenty-seven did both.
After controlling for factors such as sex, age and movements while sleeping, the researchers found that decreased slow-wave sleep coincided with higher levels of tau in the brain and a higher tau-to-amyloid ratio in the cerebrospinal fluid.
“The key is that it wasn’t the total amount of sleep that was linked to tau, it was the slow-wave sleep, which reflects quality of sleep,” Lucey said. “The people with increased tau pathology were actually sleeping more at night and napping more in the day, but they weren’t getting as good quality sleep.”
If future research bears out their findings, sleep monitoring may be an easy and affordable way to screen earlier for Alzheimer’s disease, the researchers said. Daytime napping alone was significantly associated with high levels of tau, meaning that asking a simple question – How much do you nap during the day? – might help doctors identify people who could benefit from further testing.
“I don’t expect sleep monitoring to replace brain scans or cerebrospinal fluid analysis for identifying early signs of Alzheimer’s disease, but it could supplement them,” Lucey said. “It’s something that could be easily followed over time, and if someone’s sleep habits start changing, that could be a sign for doctors to take a closer look at what might be going on in their brains.”
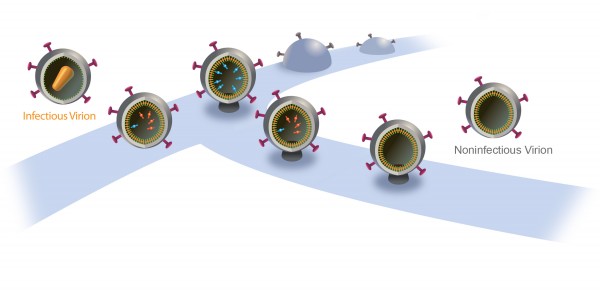
A New Way to Nip AIDS in the Bud
When new AIDS virus particles bud from an infected cell, an enzyme named protease activates to help the viruses mature and infect more cells. That’s why modern AIDS drugs control the disease by inhibiting protease.
Now, University of Utah researchers found a way to turn protease into a double-edged sword: They showed that if they delay the budding of new HIV particles, protease itself will destroy the virus instead of helping it spread. They say that might lead, in about a decade, to new kinds of AIDS drugs with fewer side effects.
“We could use the power of the protease itself to destroy the virus,” says virologist Saveez Saffarian, an associate professor of physics and astronomy at the University of Utah and senior author of the study released today by PLOS Pathogens, an online journal published by the Public Library of Science.
So-called cocktails or mixtures of protease inhibitors emerged in the 1990s and turned acquired immune deficiency syndrome into a chronic, manageable disease for people who can afford the medicines. But side effects include fat redistribution in the body, diarrhea, nausea, rash, stomach pain, liver toxicity, headache, diabetes and fever.
“They have secondary effects that hurt patients,” says Mourad Bendjennat, a research assistant professor of physics and astronomy and the study’s first author. “And the virus becomes resistant to the inhibitors. That’s why they use cocktails.”
Bendjennat adds that by discovering the molecular mechanism in which protease interacts with HIV, “we are developing a new approach that we believe may be very efficient in treating the spread of HIV.”
However, he and Saffarian emphasize the research is basic, and that it will be a decade before more research might develop the approach into news AIDS treatments.
Figuring out the role of protease in HIV budding
Inside a cell infected by HIV, new virus particles are constructed largely with a protein named Gag. Protease enzymes are incorporated into new viral particles as they are built, and are thought to be activated after the new particles “bud” out of infected cell and then break off from it.
The particles start to bud from the host cell in a saclike container called a vesicle, the neck of which eventually separates from the outer membrane of the infected cell. “Once the particles are released, the proteases are activated and the particles transform into mature HIV, which is infectious,” Saffarian says.
“There is an internal mechanism that dictates activation of the protease, which is not well understood,” he adds. “We found that if we slow the budding process, the protease activates while the HIV particle is still connected to the outer membrane of host [infected] cell. As a result, it chews out all the proteins inside the budding HIV particle, and those essential enzymes and proteins leak back into the host cell. The particle continues to bud out and release from the cell, but it is not infectious anymore because it doesn’t have the enzymes it needs to mature.”
Budding HIV needs ESCRTs
The scientists found they could slow HIV particles from budding out of cells by interfering with how they interact with proteins named ESCRTs (pronounced “escorts”), or “endosomal sorting complexes required for transport.”
ESCRTs are involved in helping pinch off budding HIV particles – essentially cutting them from the infected host cell.
Saffarian says scientific dogma long has held “that messing up the interactions of the virus with ESCRTs results in budding HIV particles permanently getting stuck on the host cell membrane instead of releasing.” Bendjennat says several studies in recent years indicated that the particles do get released, casting some doubt on the long held dogma.
The new study’s significance “is about the molecular mechanism: When the ESCRT machinery is altered, there is production of viruslike particles that are noninfectious,” he says. “This study explains the molecular mechanism of that.”
“We found HIV still releases even when early ESCRT interactions are intentionally compromised, however, with a delay,” Saffarian says. “They are stuck for a while and then they release. And by being stuck for a while, they lose their internal enzymes due to early protease activation and lose their infectivity.”
Bendjennat says by delaying virus budding and speeding “when the protease gets activated, we are now capable of using it to make new released viruses noninfectious”
How the research was done
The experiments used human skin cells grown in tissue culture. It already was known that new HIV particles assemble the same way whether the infected host cell is a skin cell, certain other cells or the T-cell white blood cell infected by the virus to cause AIDS. The experiments involved both live HIV and so-called viruslike particles.
Bendjennat and Saffarian genetically engineered mutant Gag proteins. A single HIV particle is made of some 2,000 Gag proteins and 120 copies of proteins known as Gag-Pol, as well as genetic information in the form of RNA. Pol includes protease, reverse transcriptase and integrase – the proteins HIV uses to replicate.
The mutant Gag proteins were designed to interact abnormally with two different ESCRT proteins, named ALIX and Tsg101.
A new HIV particle normally takes five minutes to release from an infected cell.
When the researchers interfered with ALIX, release was delayed 75 minutes, reducing by half the infectivity of the new virus particle. When the scientists interfered with Tsg101, release was delayed 10 hours and new HIV particles were not infectious.
The scientists also showed that how fast an HIV particle releases from an infected cell depends on how much enzyme cargo it carries in the form of Pol proteins. By interfering with ESCRT proteins during virus-release experiments with viruslike particles made only of Gag protein but none of the normal Pol enzymes, the 75-minute delay shrank to only 20 minutes, and the 10-hour delay shrank to only 50 minutes.
“When the cargo is large, the virus particle needs more help from the ESCRTs to release on a timely fashion,” Saffarian says.
Because HIV carries a large cargo, it depends on ESCRTs to release from an infected cell, so ESCRTs are good targets for drugs to delay release and let HIV proteases leak back into the host cell, making new HIV particles noninfectious, he says.
Bendjennat says other researchers already are looking for drugs to block ESCRT proteins in a way that would prevent the “neck” of the budding HIV particle from pinching off or closing, thus keeping it connected to the infected cell. But he says the same ESCRTs are needed for cell survival, so such drugs would be toxic.
Instead, the new study suggests the right approach is to use low-potency ESCRT-inhibiting drugs that delay HIV release instead of blocking it, rendering it noninfectious with fewer toxic side effects, he adds.